Dmitry Gakamskya,* and Anna Gakamskyb
aDMG Biophotonics Ltd, 1/11 Charlesfield ln, Livingston Business Centre, Livingston, EH54 7FA, UK. E-mail: dmitry@dmgbiophotonics.com
bEdinburgh Instruments Ltd, 2 Bain Square, Livingston, EH54 7DQ, UK
Fluorescence methods have found a wide range of important applications in science and technology. Compared with absorption, fluorescence gives a many-fold higher sensitivity and specificity. In addition, fluorescence possesses several important parameters such as intensity, excitation and emission spectra, polarisation, lifetime, and quantum yield. These parameters are functions of solvent nature, temperature, polarity and viscosity, and can be used to research the structure and function of various systems and for analytical applications. For example, fluorescence polarisation and non-radiative resonance energy transfer are used for detection of biomarkers in in vitro medical diagnostics.1
However, the use of fluorescence in in vivo diagnostics is hindered by the limited penetration of the visible light into living tissue or requires the use of fibre-optic probes. At the same time, proteins, being a major component of living organisms, should have diagnostic and prognostic potential.
The intrinsic fluorescence of proteins is caused by three amino acid residues with aromatic side chains: phenylalanine, tyrosine and tryptophan. Out of these three, the latter plays the most important role due to its excitation and emission spectra having the longest wavelength (near the UV range) and longest lifetime. These features simplify measurements of its fluorescence and allow for its selective detection. However, contrary to its universal nature, tryptophan fluorescence of proteins has been predominantly used only in research to study protein conformational changes or protein–protein interactions. A question arises whether protein fluorescence can be used in medical diagnostics?
Success in understanding the role of protein conformation in homeostasis of living organisms2 could provide solid support to this type of diagnostics. It has become apparent that protein post-translational modifications (PTM), causing protein mis-folding and aggregation, are responsible for a broad range of diseases, such as Parkinson’s and Alzheimer’s produced by degeneration in the brain tissue, cardiomyopathy caused by degeneration of the heart muscles or cataract caused by colouration and aggregation of the eye lens proteins.
Red-edge excitation of eye lens proteins
This generic basis for conformational disorders led us to conceive whether it would be possible to use protein fluorescence to detect changes in the eye lens structure. The eye lens created by nature is a perfectly transparent organ that represents an excellent experimental model for fluorescence measurements. A challenge in the use of tryptophan (Trp) fluorescence for cataract diagnostics is the incredibly high concentration of the eye lens proteins (crystallins) (200–400 mg mL–1) which makes the lens’ optical density in the spectral range of the Trp absorption band (260–300 nm) a hundred of units. Such a high optical density does not allow the excitation light to penetrate deeper than a hundred of microns into the lens body. However, this experimental challenge can be overcome by the use of the so-called red-edge excitation of Trp; meaning excitation on the long-wavelength (“red”) slope of the absorption band.3 First, due to the steepness of this slope, the optical density drops significantly and an excitation light of 317 nm wavelength travels throughout the lens with an attenuation of just about 25% (Figure 1, left panel).
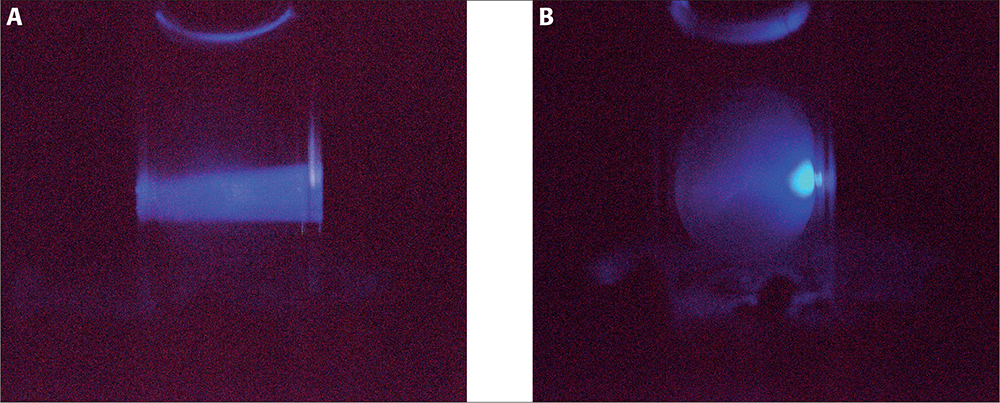
Figure 1. Excitation of eye lens fluorescence in a normal (A) and UV-irradiated (B) porcine lens by a 317 nm LED.
Second, the red-edge excitation selectively brings to the excited state a fraction of Trp side chains situated in a polar environment (“red-shifted” fraction). This fraction correlates either with all side chains of misfolded proteins exposed to an aqueous environment, or with side chains of folded proteins situated on the surface or in hydrophilic “pockets”. Hence, the combination of these two features gives the possibility to excite the “red-shifted” fraction throughout the lens.
Earlier, this method was successfully used to study the structure and function of class I Major Histocompatibility Complex proteins.4 With a view to applying this method to cataract diagnostics, we measured the fluorescence of soluble crystallins as a function of excitation wavelength. It was found that the fluorescence spectra exhibited a gradual red shift proceeding along with the shift of the excitation wavelength towards the red edge of the Trp absorption spectrum (bathochromic shift). In accord with the prediction of the red-edge model, the emission spectrum was maximally shifted to the red position when the protein was dissolved in 8 M urea, which is known to have a strong denaturing property.5 This successful result encouraged us to employ the method to a whole eye lens. We compared the red-edge fluorescence of a normal and UV-irradiated porcine lens, which was used as a model of cataract. Results of this experiment are shown in Figure 2.
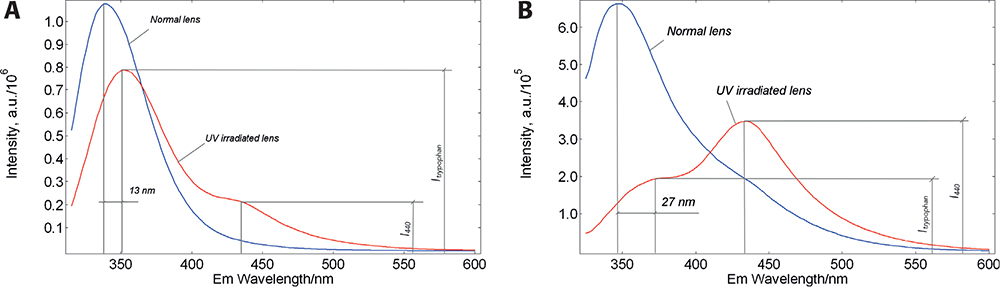
Figure 2. Red-edge excitation of eye lens proteins. Corrected fluorescence spectra of a normal and UV-irradiated porcine lens measured in a FLS980 spectrometer in 90° geometry. Excitation at 305 nm (A) caused a red shift of Trp emission by 13 nm and the appearance of a low-intensity, additional emission band at 440 nm (I440 / ITrp < 1). While upon excitation at 315 nm (B) the Trp emission peak is shifted by 27 nm and the relative intensity of the addition band is increased (I440 / ITrp > 1).
In part, these results are consistent with the model, as the emission band of the UV-irradiated lens showed a greater amplitude red-shift. However, there is a significant difference: the emission spectra of the normal and irradiated lenses show an unexpected additional band on the red slope of the emission spectrum. The UV irradiation decreased the amplitude of the Trp emission band and increased the intensity of the additional band. This situation seems to indicate that the red-shifted fraction of Trp converts to a newly formed fluorophore with emission maximum at ~440 nm. It was also found that the ratio of the 440 nm band to the Trp band positively correlates with the irradiation dose5 and the sensitivity of this ratiometric parameter is much greater than the sensitivity of the slit-lamp method of light scattering. To be able to use this additional fluorescence band for diagnostic purposes, it is important to understand its chemical nature.
Decomposition: a key approach for spectra analysis
Mass spectrometry of irradiated porcine lens solubilised proteins and solubilised human post-operational protein samples revealed that, together with tryptophan residues, these proteins contain two primary products of Trp: hydroxytryptophan (OH-Trp) and N-formylkynurenine (NFK) and two downstream products, kynurenine (Kyn) and hydroxykynurenine (OH-Kyn). In addition, a fluorescent derivative of arginine, argpyrimidine (ArgP), was identified.6 Importantly, all the fluorophores could be excited by the same wavelength of 317 nm, which was earlier found as a trade-off wavelength for deep lens penetration and Trp excitation.
To make this method semi-quantitative, we need to normalise the non-Trp emission against a reference signal. This signal should vary to the same extent as the emission of the fluorescent PTMs as a function of experimental conditions (intensity of excitation, geometry etc.). Since the concentration of Trp remains nearly constant in spite of the modifications (1–2%), emission of its “red-shifted” fraction ideally fits this purpose.
Knowledge of the spectral properties of each fluorescence component greatly simplifies calculations of concentration of the individual components, which can be determined by decomposing the total emission spectrum. Figure 3 shows a typical example of the spectral decomposition emission spectrum measured from a post-operational emulsified sample with the second grade of nuclear cataract (NC++). The quality of the decomposition given by the shape and amplitude of the residuals function suggests that the elementary spectral components were correctly determined. We found that, for a statistically representative number of experimental samples, emission of OH-Trp, NFK and ArgP comprised the major portion of the total emission spectra. Assuming that the accumulation of PTMs causes cataractogenesis, we correlated the cumulative contribution of these three components with the grade of cataract measured by a slit-lamp.6 The data revealed a Pearson’s coefficient of correlation of 0.93. A smaller (0.68), but still significant, coefficient was found for the correlation of the cumulative emission of these fluorescent modifications with donors’ age for 17 post-mortal lenses. Thus, this method can be used for a non-invasive, semi-quantitative cataract diagnosis, with greatly increased sensitivity over current techniques.
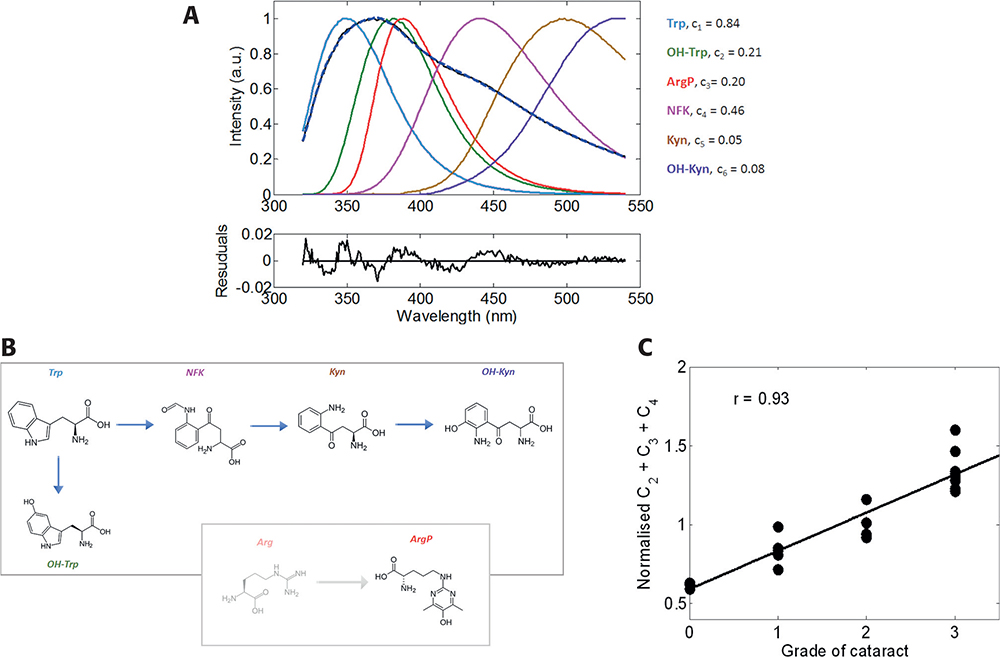
Figure 3. Fluorescent spectrum of the insoluble fraction of emulsified cataractous sample (A, black) and its decomposition determined from spectra of identified fluorescent derivatives of Trp and ArgP (B). Correlation of cataract grade with the normalised cumulative fraction for 21 emulsified eye lens proteins (C).
A wide range of PTMs in crystallins was identified by mass spectrometry and it is likely that all modifications may contribute to protein degradation. A question therefore arises—why does the fluorescent fraction, from all possible PTMs, correlate with cataract grade and age? While the significance of every PTM is not clearly understood, we hypothesise that the fluorescent fraction represents the total modification statistics. Moreover, some specific fluorescent PTMs has been shown to play important roles in cataractogenesis by changing the stability and interactions of proteins. Ghosh et al.7 demonstrated that the interaction between the N-terminal W60 and the core domain R123 plays a role in the monomeric interaction of α-crystallins involved in the formation of the α-crystallin oligomeric structure. The assembly of α-crystallins into large oligomeric complexes is believed to be central to the chaperone-like function of this protein.
The formation of NFK from Trp is mediated by the interaction with singlet oxygen and, therefore, the concentration of NFK can be used as a marker of protein oxidative stress. Moreover, NFK possesses photosensitising properties, which make it the most biologically important photo-product of Trp because its formation facilitates the production of hydrogen peroxide in irradiated Trp solutions. Hence, the formation of NFK can stimulate modifications of other side chains.
We found abundant formation of ArgP in α-crystallins and, in particular, the conversion of Arg120 to ArgP in α-crystallin B is likely to affect its chaperone-like function. Vicart et al.8 identified the Arg120Gly mutation in α-crystallin B as responsible for desmin-related myopathy in a French family. It has also been shown that cataract and myopathy pathologies in α B-Arg120Gly knock-in mice share common mechanisms, and mutation of the Arg120 residue in the human α-crystallin causes a partial loss of its chaperone-like activity. All these findings strongly suggest that the concentration of Arg120 should negatively correlate with the chaperone-like function of α-crystallin and hence may be used as a marker of cataract.
In summary, we discussed here a novel, non-invasive method of semi-quantitative determination of the concentrations of fluorescent PTMs in the lens based on simultaneous measurements of the fluorescence emission spectra of Trp, its photo-degradation products and ArgP, which can be used for diagnostics of cataract at the molecular level. This makes the method useful for cataract grading and for monitoring cataractogenesis over a period of time. The latter may help in elucidation of various metabolic and ambient factors contributing to cataractogenesis. The possibility to monitor changes in the lens structure at the molecular level may also facilitate the development of cataract medications, aimed at slowing down the cataractogenic processes and prolonging lens homeostasis.
References
- D. Wild (Ed), The Immunoassay Handbook Theory and Applications of Ligand Binding, ELISA and Related Techniques. Elsevier (2013).
- R.J.W. Truscott, in Protein Misfolding, Aggregation, and Conformational Diseases, Ed by V.N. Uversky and A.L. Fink. Springer, pp. 436–447 (2007).
- A.P. Demchenko, Ultraviolet Spectroscopy of Proteins. Springer (1986). doi: https://doi.org/10.1007/978-3-642-70847-3
- D.M. Gakamsky, E. Haas, P. Robbins, J.L. Strominger and I. Pecht, “Selective steady-state and time-resolved fluorescence spectroscopy of an HLA-A2-peptide complex”, Immunol. Lett. 44, 195–201 (1995). doi: https://doi.org/10.1016/0165-2478(94)00214-C
- D.M. Gakamsky, B. Dhillon, J. Babraj, M. Shelton and S.D. Smith, “Exploring the possibility of early cataract diagnostics based on tryptophan fluorescence”, J. R. Soc. Interface 8, 1616–1621 (2011). doi: https://doi.org/10.1098/rsif.2010.0608
- A. Gakamsky, R.R. Duncan, N.M. Howarth, B. Dhillon, K.K. Buttenschön, D.J. Daly and D. Gakamsky, “Tryptophan and non-tryptophan fluorescence of the eye lens proteins provides diagnostics of cataract at the molecular level”, Sci. Reports 7, 40375 (2017). doi: https://doi.org/10.1038/srep40375
- J.G. Ghosh and J.I. Clark, “Insights into the domains required for dimerization and assembly of human aB crystallin”, Protein Sci. 14, 684–695 (2005). doi: https://doi.org/10.1110/ps.041152805
- P. Vicart, A. Caron, P. Guicheney, Z. Li, M.-C. Prévost, A. Faure, D. Chateau, F. Chapon, F. Tomé, J.-M. Dupret, D. Paulin and M. Fardeau, “A missense mutation in the αB-crystallin chaperone gene causes a desmin-related myopathy”, Nat. Genet. 20, 92–95 (1998). doi: https://doi.org/10.1038/1765