
Michael J. Watts,a* Mark Buttona,b and Gawen R.T. Jenkinb
aBritish Geological Survey, Nottingham, NG12 5GG, UK. E-mail: mwatts@bgs.ac.uk
bDepartment of Geology, University of Leicester, Leicester, UK
Introduction
The health implications of chronic exposure to arsenic are well known, with populations exposed on a worldwide scale, the majority of which are in locations such as Bangladesh, South-East Asia and South America. Serious health problems have been associated with drinking water high in arsenic, including various cancers, vascular disease and skin keratoses.1 In the UK, arsenic concentrations in drinking water are strictly regulated2 to meet the World Health Organisation recommended upper limit of 10 µg L–1 As for drinking water.3 Significant exposure can also arise from the consumption of contaminated food, inhalation of dust or ingestion of contaminated soil. The UK region Devon and Cornwall, which are located in the south-west of England, have a historical legacy of past mining activities which has led to locations with elevated levels of arsenic in soils, where soil ingestion is expected to be a significant exposure risk.4 Increasing evidence of exposure in these areas5,6 has resulted in growing concern about the potential health effects in populations exposed to arsenic. The assessment of exposure is inherently complex, in that the total amount of arsenic in soil provides an indication of the extent of the problem, but not the direct risk from exposure. The toxicity of arsenic is largely species dependent, with several inorganic and organic species being highly toxic7 yet other common species such as arsenobetaine and arsenocholine have been reported to be of minor toxicological significance.8 The speciation of arsenic is therefore important in understanding the pathways from source to exposure, biotransformation following exposure and associated risk to the health of organisms and humans.
Ecosystem indicator species such as earthworms have been employed as a tool for assessing soil contamination, in particular using bioaccumulation as a guide to a contaminant bioavailability.9,10 Whilst not directly indicating potential human health effects, the use of sentinel organisms such as earthworms may provide a useful surrogate to human studies at arsenic contaminated sites and provide a complementary line of evidence in assessing risk.9 However, biomonitoring of human populations is needed to provide quantitative estimates of exposure to arsenic. Urine is often used, although the residence time of arsenic is about 24 hours11 meaning that it will most likely be influenced by dietary intake and is not ideal for assessing exposure from soil where ingestion may be sporadic. Long-term measures of exposure including hair and nails provide a time-integrated sample for assessing the uptake of arsenic in humans.
Analytical methodologies were developed for the speciation of arsenic in a range of materials by coupling high performance liquid chromatography with inductively coupled plasma mass spectrometry (HPLC-ICP-MS) to understand the exposure and uptake of arsenic in bioindicator organisms and humans. This work was part of a British Geological Survey sponsored PhD studentship, in collaboration with the University of Leicester. This article highlights the versatility of the developed methodology for the measurement of arsenic species in a range of materials from Devon Great Consols (DGC), one of many former mining sites in the south- west of England. A summary of results is presented with a focus on the instrumental measurement of arsenic species, whilst greater detail regarding sampling sites, sample preparation for analysis, and detailed interpretation of the findings can be found in the appropriate publications for this work.6,9,12–14
Methods
Samples
Earthworms and host soils for analysis were collected from both the contaminated site at DGC and from an uncontaminated area in Nottingham.9 Toenail samples were obtained from both residents of properties near the former mining site of DGC and from volunteers in the Nottingham area.6
Extraction
The extraction and measurement of arsenic species was monitored using certified reference material CRM 627 tuna fish for earthworm analyses, NCS ZC 81002b human hair (NCS Beijing, China) and CRM 13 human hair (NIES, Onogwa, Japan) for toenail analyses. Prior to extraction, the stability of arsenic species AsIII (arsenite), AsV (arsenate), MAV (methylarsonate), DMAV (dimethylarsinate) and AB (arsenobetaine) was established under the extraction conditions employed by spiking each of the earthworm powder materials with each of the arsenic species. Further detail for certified reference material and spike recovery data, including extraction and total digestion methodologies are discussed in detail elsewhere.6,13,14
Instrumentation
During the course of this work two ICP-MS instruments were used: initially for earthworm analyses, a Thermo Elemental PQ ExCell (Thermo Scientific, UK), and also for earthworms and toenails, an Agilent 7500 ICP-MS (Agilent Technologies, UK). Both instruments were fitted with microconcentric nebulisers, the former with an impact bead spray chamber and the latter with a Scott double pass spray chamber. The instrument response was optimised using a 1 µg L–1 Claritas multielement tune solution 1 (GlenSpectra, UK). Indium (10 µg L–1) and Te (50 µg L–1) were used as internal standards, added to the sample stream via a T-piece. The Agilent 7500 employed a collision cell using He (5.5 mL min–1) to minimise potential interferences such as that from the polyatomic ion Ar40Cl35.
A quaternary pump (GP-50) and AS-50 autosampler (Dionex, USA) were directly coupled to the ICP-MS for measurement of arsenic species via the analytical column, which was connected to the ICP nebuliser using PEEK tubing. The two instruments were coupled in such a way that the injection of each sample solution via the autosampler and subsequent measurement was synchronised automatically, enabling reproducible sample injections. Two eluent systems were used for the separation of extracted arsenic species present in earthworms and toenails. These were an anion exchange column with a guard cartridge of the same material or a cation exchange column with a guard cartridge of the same material.12,14 The first chromatographic separation (Figure 1) was based on a modified gradient elution system developed by Martinez-Bravo et al.15 which used a gradient between A: 4 mM NH4NO3 and B: 60 mM NH4NO3, both adjusted to pH 8.7 with ammonia. This method14,15 achieved good separation for AsIII, AsV, MA, DMA, and the phosphate, sulphonate and sulphate arsenosugars. Arsenosugars were only identified in earthworms and not in soils or toenails. Poor separation was achieved for AB and the glycerol arsenosugar. Therefore a second chromatographic separation (Figure 2) was employed, which was a modified version of an isocratic method developed by Geiszinger et al.,16 for the measurement of AB and the glycerol arsenosugar. The cation exchange system used 10 mM pyridine at pH 2.26, adjusted with formic acid and an isocratic flow rate of 1.5 mL min–1. An injection volume of 100 µL was used for both systems. Peak areas from the PQ ExCell ICP-MS were calculated using PeakFit 4.0 (Seasolve Software, USA) and for the Agilent ICP-MS with Chemstation instrument software. Quantified data was produced by inclusion of blank (deionised water) and calibrant standards for each arsenic species from 2 µg L–1 to 50 µg L–1 in each analytical run. Isolated arsenosugar standards were utilised for the identification of arsenosugars by retention time matching, whilst the calibration for MA was for the quantification of the arsenosugars. The gradient elution method (anion exchange) provided limits of detection for solutions, based on 3s for blank values for the PQ ExCell ICP-MS instrument as follows: AB: 1.3 µg L–1, AsIII: 0.8 µg L–1, AsV: 1.5 µg L–1, MA: 0.7 µg L–1, DMA: 0.3 µg L–1.
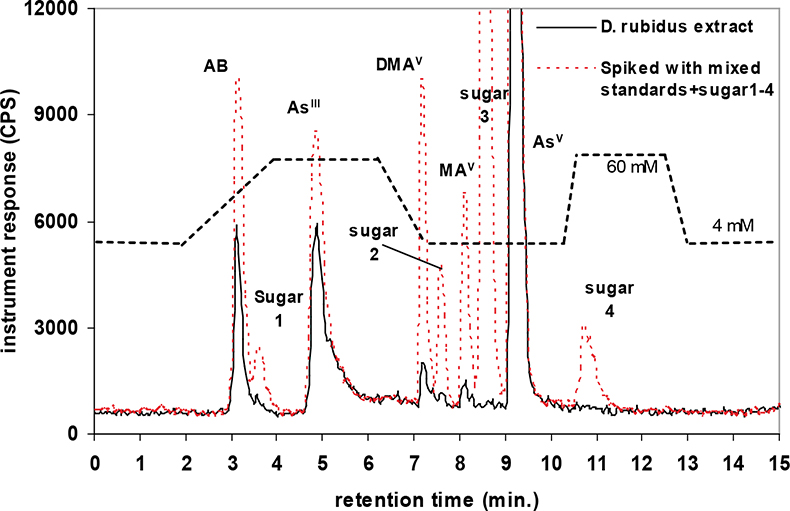
Figure 1. HPLC-ICP-MS: anion gradient elution profile for a mixed standard solution (10 µg L–1) and a representative earthworm extract. The dashed line indicates transitions in the mobile phase.
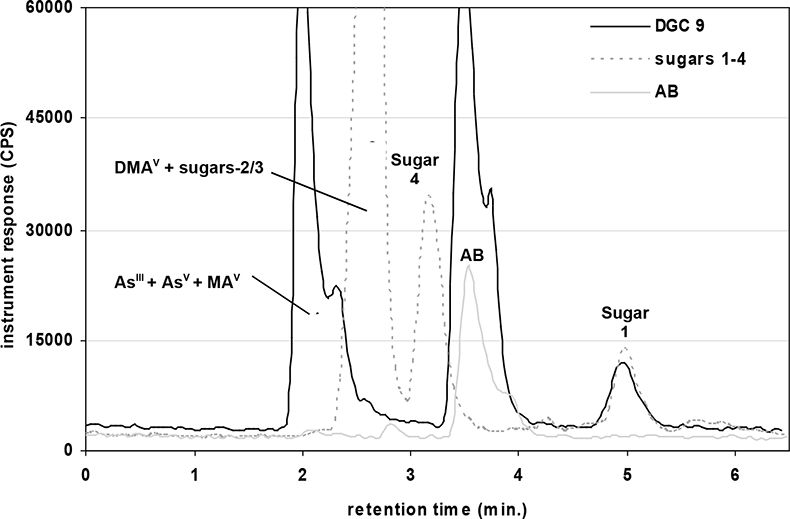
Figure 2. HPLC-ICP-MS: cation isocratic elution profile for an earthworm extract (D. rubidus), an algal extract containing sugars 1–4 and an AB standard at 13 µg L–1.
Results
Table 1 summarises the arsenic concentrations measured and the proportion of arsenic present as each arsenic species in the sample extract for each matrix, including soil, earthworms and toenails. Soil arsenic was found to be mainly AsV, with a small proportion of AsIII, with no other constituents detected using the anion exchange gradient elution HPLC-ICP-MS method. Arsenic was generally present in earthworms as AsV or AsIII and AB. MA, DMA and three arsenosugars were present as minor constituents. Whilst the inorganic arsenic increased with increasing arsenic body burden, the concentration of organic species remained relatively constant.12 Total arsenic concentrations in toenail samples were positively correlated to environmental arsenic levels (r = 0.60, p < 0.001).12 Arsenic resided in toenails from both control and exposed groups as mainly AsIII or AsV, with DMA as a minor constituent.
Table 1. Summary of arsenic concentrations for the Devon Great Consols, arsenic species presented as a percentage of the total arsenic extracted. Range and (mean) values given.
Sample matrix | Total As (mg kg–1) | Extraction efficiency (%) | AsIII | AsV | DMAV | MAV |
Host soil (n = 23) | 255–13,080 (2615) | 70–108 (81) | 1–14 (3) | 80–113 (95) | nd | nd |
*Earthworms (n = 23) | 11–877 (256) | 27–82 (52) | 5–61 (29) | 7–84 (40) | 0.1–23 (3.6) | 0.1–5 (1.2) |
Toenails: exposed | 2987–25981 | 55–68 | 81–86 | 5–21 | 0.5–2 | nd |
nd: below limits of detection
*Earthworms: arsenosugars 1 (glycerol): 0.01–6 (1.4%), 2 (phosphate): 0.04–5 (1.36%), 4 (sulphate): 0.1–0.8 (0.31%)
Conclusion
A gradient elution anion exchange method that required the coupling of an HPLC to an ICP-MS instrument was utilised for the measurement of nine arsenic species from one sample injection. This method was effectively employed for the arsenic speciation of a range of matrices and helps to better understand the pathway of exposure-uptake, biotransformation of arsenic following exposure to arsenic and subsequent body burden within earthworms. This information provides new insight into determining the arsenic exposure, risk and cycling in the environment when using earthworms as a bioindicator organism. This method also provided arsenic speciation data for the distribution of arsenic in toenails, with arsenic mainly residing as AsIII in both the exposed and control groups, with AsV and DMAV present in lower amounts. This data demonstrated the sensitivity of the toenails as a biomarker of exposure to high environmental arsenic, resultant from historical mining activities. Further studies are on-going to explore the environmental exposure of arsenic applying this methodology to measure arsenic species in drinking water and other relatively non-invasive human biomarkers of exposure, including hair and urine to better understand the fate and metabolism of arsenic.
References
- M.R. Karagas, T.A. Stukel and T.D. Tosteson, Int. J. Hyg. Environ. Health. 205, 85–94 (2002). https://doi.org/10.1078/1438-4639-00133
- DWI, Drinking Water Inspectorate, London (2008).
- World Health Organisation, Guidelines for Drinking-Water Quality (1993). http://www.who.int/water_sanitation_health/dwq/arsenicsum.pdf
- A.G. Oomen, A. Hack, M. Minekus, E. Zeijdner, C. Cornelis, G. Schoeters, W. Verstraete, T. Van De Wiele, J. Wragg, C.J.M. Rompelberg, A.J.A.M. Sips and J.H. Van Wijnen, Environ. Sci. Technol. 36, 3326–3334 (2002). https://doi.org/10.1021/es010204v
- P. Kavanagh, M.E. Farago, I. Thornton, W. Goessler, D. Kuehnelt, C. Schlagenhaufen and K.J. Irgolic, Analyst 123, 27–29 (1998). https://doi.org/10.1039/a704893i
- M. Button, G.R.T. Jenkin, C.F. Harrington and M.J. Watts, J. Environ. Monitor. 11, 610–617 (2009). https://doi.org/10.1039/b817097e
- K.T. Kitchin, Toxicol. Appl. Pharmacol. 172, 249–261 (2001). https://doi.org/10.1006/taap.2001.9157
- T.W. Gebel, Int. J. Hyg. Environ. Health 203, 249–262 (2001). https://doi.org/10.1078/S1438-4639(04)70036-X
- M. Button, M.J. Watts, M. Cave, C.F. Harrington and G.R.T. Jenkin, Environ. Geochem. Health 31, 273–282 (2009). https://doi.org/10.1007/s10653-008-9208-3
- C.J. Langdon, T.G. Piearce, A.A. Meharg and K.T. Semple, Environ. Pollut. 124, 361–373 (2003). https://doi.org/10.1016/S0269-7491(03)00047-2
- M.J. Slotnick, J.R. Meliker, G.A. Avruskin, D. Ghosh and J.O. Nriagu, J. Toxicol. Environ. Health A 70, 148–158 (2007). https://doi.org/10.1080/15287390600755232
- Button, G.R.T. Jenkin, C.F. Harrington and M.J. Watts, J. Environ. Monit. in press (2009). https://doi.org/10.1039/b904104d
- Button and M.J. Watts, British Geological Survey IR/08/050 (2008).
- M.J. Watts, M. Button, T.S. Brewer, G.R.T. Jenkin and C.F. Harrington, J. Environ. Monitor. 10, 753–759 (2008). https://doi.org/10.1039/b800567b
- Y. Martinez-Bravo, A.F. Roig-Navarro, F.J. Lopez and F. Hernandez, J. Chromatogr. A 926, 265–274 (2001). https://doi.org/10.1016/S0021-9673(01)01062-7
- Geiszinger, W. Goessler and W. Kosmus, Appl. Organomet. Chem. 16, 473–476 (2002). https://doi.org/10.1002/aoc.327







